- SPECTROCHIMIQUE (ANALYSE)
- SPECTROCHIMIQUE (ANALYSE)Tous les éléments chimiques peuvent en principe être identifiés par la présence de raies caractéristiques dans leur spectre d’émission d’arc ou d’étincelle, ou encore dans le spectre d’émission de rayons X (cf. chimie ANALYTIQUE).Pour identifier les molécules ou des groupes fonctionnels de composés organiques ou minéraux, on a recours à de nombreuses méthodes spectroscopiques d’analyses dont les caractéristiques principales les plus utilisées sont résumées dans le tableau 1. Leurs apports respectifs à la résolution de problèmes de structure seront successivement abordés. On utilise aussi les spectroscopies de résonance quadrupolaire nucléaire (R.Q.N.), d’absorption électronique, d’absorption atomique, de micro-ondes et Mössbauer.La plupart des méthodes spectroscopiques reposent sur un principe commun: une source d’énergie fournit une onde électromagnétique qui est dirigée sur la substance à étudier. Suivant le domaine de longueur d’onde utilisé, cette radiation est diffractée par les plans de réflexion du cristal (rayons X), ou absorbée par le cristal ou la solution (U.V., I.R., R.M.N., R.P.E.). Dans le premier cas, les caractéristiques de la lumière diffractée permettent de localiser les atomes à l’intérieur de la maille cristalline. Dans le second cas (spectroscopie d’absorption), l’absorption variera avec la longueur d’onde utilisée: l’intensité et la position de l’absorption dans le spectre pourront donc être mises en corrélation avec des caractéristiques structurales de la molécule et permettront ainsi de localiser les atomes les uns par rapport aux autres à l’intérieur d’une molécule. Bien souvent ces méthodes fournissent aussi des indications précieuses sur les caractéristiques des liaisons interatomiques – géométrie, force, polarité – et permettent ainsi d’interpréter ou de prévoir la réactivité des molécules.Pour un composé, des différences significatives peuvent exister entre sa structure à l’état solide et sa structure en solution (cf. état LIQUIDE): la première est déterminée essentiellement au moyen de la diffraction des rayons X, et la stabilité conformationnelle de la molécule dépendra non seulement des forces intramoléculaires, mais aussi des forces cristallines. Par contre, la conformation d’une molécule en solution sera influencée par les interactions solvant-soluté et soluté-soluté, dont l’importance sera elle-même fonction de différentes grandeurs physiques: température, concentration, polarité du solvant et du composé dissous, nature des autres substances présentes en solution, etc. Pour ces raisons, un composé peut adopter plusieurs conformations différentes en solution. Notons que certaines méthodes spectroscopiques, la spectroscopie de micro-ondes et la R.M.N. par exemple, permettent non seulement de préciser les conformations mais aussi d’étudier ces mouvements de rotation ou de translation dont une molécule peut être le siège.Les progrès technologiques accomplis ces dernières années se sont traduits en particulier par l’adjonction aux spectrographes de micro-ordinateurs permettant leur pilotage automatique ainsi qu’un traitement rapide de l’information et un stockage des résultats expérimentaux. Ils ont aussi rendu possible l’analyse structurale de molécules de plus en plus complexes: protéines, acides nucléiques ou polysaccharides, à partir de quantités minimes de produits (de l’ordre de quelques milligrammes).1. Structure des composés organiquesAfin de préciser l’utilité et les limites d’application des méthodes spectroscopiques pour les composés organiques, on développera leur application à l’analyse structurale des oligopeptides.Les peptides – cf. la formule générale (1) – sont des constituants de la matière vivante résultant de la combinaison d’acides aminés reliés entre eux par une liaison peptidique 漣 C(= O) 漣 NH 漣, formée par l’élimination d’une molécule d’eau entre le groupement carboxyle 漣 COOH de l’un et le groupement amino 漣 NH2 de l’autre. Ces peptides peuvent être présents à l’état naturel: c’est le cas des hormones comme la thyroxine ou l’angiotensine. On les obtient également par dégradation des protéines ou par synthèse chimique. L’étude de leur structure est un problème chimique et biochimique important, souvent rendu difficile par les faibles quantités de produits disponibles.Les oligopeptides diffèrent les uns des autres par la nature des acides aminés présents, par leur nombre, par leur séquence dans la chaîne peptidique. L’analyse spectrochimique contribue à résoudre ces trois problèmes (structure primaire) et permet en outre le plus souvent de déterminer la structure spatiale de ces molécules.Diffraction des rayons XL’analyse structurale de plusieurs peptides à l’état cristallin a été effectuée au moyen de la diffraction des rayons X. Les longueurs des liaisons entre les atomes sont déterminées avec une erreur expérimentale souvent inférieure à 0,0002 nm.Ces études ont permis de montrer que les atomes des groupements de la formule (2) étaient coplanaires, confirmant ainsi une hypothèse formulée en 1953 par L. Pauling et R. B. Corey. D’autre part, les peptides de petite dimension adoptent à l’état solide des conformations «pelotes statistiques» [cf. ALCANES] et non une structure secondaire bien définie (hélices 見 et/ou feuillets 廓) comme c’est le cas pour les peptides plus grands ou dans les protéines [cf. PROTÉINES].Au cours des dix dernières années, le développement considérable de cette méthode d’analyse a permis de déterminer la structure tridimensionnelle à haute résolution de plusieurs centaines de macromolécules biologiquement importantes. Les informations structurales ainsi recueillies ont apporté une meilleure connaissance du mécanisme d’action de ces composés et ont permis, par la suite, la mise au point de nombreux produits pharmacologiques.La diffraction des neutrons est une méthode voisine également utilisée.Spectroscopie ultraviolette et visibleL’absorption de lumière ultraviolette par une molécule organique induit des transitions électroniques entre des états d’énergie stables et des états d’énergie excités. La longueur d’onde de cette absorption est dans une certaine mesure caractéristique des groupements fonctionnels responsables de ces transitions.Le spectre d’absorption ultraviolette de peptides varie avec la nature des acides aminés qui les composent: si ceux-ci ne contiennent pas de noyaux aromatiques dans leur chaîne latérale, le spectre comporte un seul maximum vers 195-200 nm, correspondant à la liaison peptidique. Un deuxième maximum est observé à une longueur d’onde plus élevée lorsque le peptide contient au moins un acide aminé à chaîne latérale aromatique. Ce maximum est de faible intensité pour un résidu phénylalanine, d’intensité plus forte pour des résidus tryptophane et tyrosine (fig. 1). Pour cette dernière, la position du maximum varie lorsque la solution est rendue basique, ce qui permet l’identification de cet acide aminé.La spectroscopie ultraviolette est une méthode très sensible qui nécessite de faibles quantités de produit (face=F0019 黎 0,5 mg) et qui conduit à l’identification facile des résidus d’acides aminés à chaîne latérale aromatique.Polarimétrie, dispersion rotatoire optique et dichroïsme circulaireLa lumière polarisée plane peut être décrite comme étant la somme de deux composantes polarisées circulairement droite et gauche d’égale intensité. Toute substance optiquement active, comme un acide aminé (sauf la glycine) dont l’activité optique provient de la présence du carbone asymétrique, interagit différemment avec chacune de ces deux composantes. Il en résulte une rotation du plan de la lumière polarisée plane d’un angle 見 appelé pouvoir rotatoire .La polarimétrie permet de déterminer le pouvoir rotatoire 見 d’une substance à une longueur d’onde donnée de la lumière monochromatique utilisée pour la mesure. La dispersion rotatoire optique est la variation de 見 en fonction de . Le dichroïsme circulaire est lié à la dispersion rotatoire optique, mais mesure l’ellipticité de la lumière émergente en fonction de , provenant de la différence des coefficients d’extinction molaires de la substance vis-à-vis de la lumière polarisée circulairement droite et de celle qui est polarisée circulairement gauche. Si l’ellipticité varie au voisinage du maximum d’absorption d’un groupement fonctionnel présent dans la molécule, par exemple la liaison peptidique entre 200 et 230 nm, la courbe prend une forme caractéristique (fig. 2) qui traduit l’effet Cotton du groupement en question. Le signe et la valeur de cet effet changent avec la conformation adoptée par les oligopeptides (l’essentiel de leur activité optique résulte des conformations qu’ils peuvent prendre, et peu de l’asymétrie des aminoacides qui les constituent). Les courbes sont suffisamment différentes les unes des autres pour permettre de préciser leur conformation, y compris celle de protéines. On utilise souvent les spectres de dichroïsme circulaire des protéines au niveau de la bande d’absorption de la liaison peptidique afin d’estimer la proportion des aminoacides qui ont une conformation hélice 見, feuillet 廓 ou pelote statistique. Le dichroïsme circulaire est également une méthode de choix pour déterminer les changements de conformations des protéines, notamment leur dénaturation.Spectroscopie infrarouge et spectroscopie RamanLorsqu’une radiation infrarouge traverse une molécule, on constate pour certaines longueurs d’onde une absorption sélective de la lumière correspondant aux fréquences de vibration caractéristiques des différentes liaisons chimiques [cf. INFRAROUGE]. Le spectre d’absorption infrarouge d’un composé est donc constitué d’un certain nombre de bandes dont l’identification fournit des renseignements précis sur la structure de ce produit.Dans le cas des peptides, deux régions du spectre sont particulièrement utiles à cet égard:– vers 3 300-3 500 cm size=1漣1 apparaissent les bandes associées aux groupements NH de la liaison peptidique. La position précise de la bande permet de savoir si le groupement est lié ou non au groupement carbonyle d’une autre liaison peptidique de la même molécule par une liaison hydrogène intramoléculaire;– entre 1 630 et 1 710 cm size=1漣1, on trouve des bandes intenses provenant du groupement carbonyle de la liaison peptidique. La fréquence de vibration de cette bande fournit des renseignements sur le degré d’hélicité de la chaîne peptidique.Contrairement à la spectroscopie infrarouge, la spectroscopie Raman n’exploite pas la polarité des liaisons chimiques, mais leur polarisabilité (cf. effet RAMAN: on évite ainsi l’absorption de l’eau gênante lors de l’étude infrarouge des peptides en solution aqueuse. Spectroscopie infrarouge et spectroscopie Raman fournissent des informations structurales tout à fait complémentaires.L’apport récent de la transformée de Fourier à ces techniques permet dorénavant l’étude de solutions relativement peu concentrées. De nouvelles méthodes d’analyse infrarouge dites «de réflexion», telles la réflexion totale atténuée (A.T.R.) et la réflexion diffuse permettent d’obtenir un spectre infrarouge de substances pures sans les matrices usuelles (KBr, Nujol, solvants) ou de composés en solution aqueuse sans être gêné par l’absorbance de l’eau et de substances diverses et exotiques (cheveu, plastiques, etc.).Spectroscopie de fluorescenceLorsqu’une molécule absorbe l’énergie d’une onde électromagnétique, elle se trouve alors dans un état électronique excité instable à température ambiante. La perte d’énergie accompagnant le retour à l’état électronique fondamental peut s’effectuer selon plusieurs processus différents, dont celui d’émission de radiations appelé fluorescence . Il n’existe qu’un nombre limité de composés fluorescents (les fluorophores). Ce sont typiquement des molécules possédant un système conjugué important d’électrons 神, comme les dérivés d’hydrocarbures aromatiques, et dont les mouvements de rotation interne ne sont pas possibles.Les oligopeptides et les protéines possédant de tels groupements (tryptophane, phénylalanine et tyrosine) sont souvent étudiés par spectroscopie de fluorescence. Les spectres obtenus fournissent des informations caractéristiques de la structure locale des fluorophores en question, ainsi que sur leur environnement (hydrophobe ou hydrophile, etc.). Au cas où les systèmes étudiés ne possèdent pas de tels groupements, on peut parfois employer des fluorophores extrinsèques, comme le bromure d’éthidium dans l’étude fluorimétrique des acides nucléiques (ADN, ARN).La fluorimétrie (la mesure de l’intensité de l’émission de fluorescence) est une technique puissante pour l’analyse quantitative. En effet, la spectroscopie de fluorescence est beaucoup plus sensible que la spectroscopie d’absorption (de 102 à 104 fois plus sensible). Dans certains cas, des concentrations inférieures à 10–12 mole/l peuvent être détectées.Résonance magnétique nucléaireLa résonance magnétique nucléaire permet en particulier l’étude de l’environnement de noyaux possédant un moment magnétique non nul: 1H, 13C, etc. [cf. RÉSONANCE MAGNÉTIQUE].Placé dans un champ magnétique, un noyau d’hydrogène peut occuper deux niveaux d’énergie entre lesquels des transitions peuvent être induites par irradiation. L’absorption de cette énergie par le proton se traduit par un signal de résonance dont la position (le déplacement chimique , exprimé en ppm) est caractéristique de l’environnement du proton en question. On observe ainsi autant de signaux que de protons ayant des environnements chimiques différents. Chaque zone de déplacements chimiques correspond à un type de proton bien précis. Les intensités relatives des signaux sont proportionnelles au nombre de noyaux, ce qui facilite encore leur identification.La multiplicité du signal d’un proton permet d’autre part d’avoir des renseignements sur le nombre de protons portés par les atomes de carbone voisins. Les espacements entre les raies d’un multiplet sont égaux aux constantes de couplage du proton correspondant (exprimées en hertz). Les valeurs de ces constantes sont fonction de la géométrie des spins considérés et fournissent donc des renseignements structuraux importants.En plus des spectres de R.M.N. de protons, ceux de carbone 13 sont souvent utilisés pour étudier les structures moléculaires. La difficulté majeure rencontrée pour obtenir de bons spectres de R.M.N. de 13C provient de la faible abondance naturelle de cet isotope (1,1 p. 100). À concentrations de produit égales, les signaux de 13C sont environ six mille fois moins intenses que ceux de proton. Pour remédier à cet inconvénient, on a développé une autre technique appelée R.M.N. par impulsions et transformée de Fourier , qui permet de recueillir l’ensemble des données expérimentales: déplacements chimiques, intensités, constantes de couplage dans un temps très bref de l’ordre de quelques secondes. On peut alors mesurer un très grand nombre de fois le même spectre et stocker les informations recueillies dans un mini-ordinateur. En effectuant la somme de tous ces spectres en fin d’expérience, on obtient un spectre de R.M.N. de 13C utilisable dont on déduit, comme d’un spectre de proton, les renseignements structuraux souhaités.Tous les autres noyaux pourvus de spins (2H, 15N, 19F, 31P, etc.), même en faible abondance isotopique, sont aujourd’hui utilisés en R.M.N. pour des études bien précises.À titre d’exemple simple, les figures 3 a et 3 b montrent respectivement les spectres de R.M.N. de 1H et de 13C (spectres dits à une dimension ou 1 D) d’un tripeptide: le glutathion ( 塚-Glu-Cys-Gly). En analysant ces spectres, on établit ou on confirme la nature des acides aminés constituant le peptide, mais on n’obtient pas sa séquence.Dans le cas de peptides plus importants et de petites protéines, l’interprétation des spectres 1 D devient vite compliquée, sinon impossible. La R.M.N. à hauts champs magnétiques, multi-impulsionnelle et à transformée de Fourier, répond à ce problème grâce au développement phénoménal des techniques d’analyse R.M.N. multidimensionnelles (2 D, 3 D, etc.). Connaissant leur séquence, on détermine désormais par R.M.N. la structure tridimensionnelle de protéines en solution contenant jusqu’à cent cinquante aminoacides! Les mouvements internes d’une protéine peuvent également être étudiés, de même que les interactions entre protéines et plus petites molécules (inhibiteurs, etc.).Les possibilités d’analyse offertes par la R.M.N. sont très nombreuses, notamment pour l’imagerie médicale.Spectrométrie de masseLe bombardement par des électrons d’une molécule organique à l’état gazeux a pour effet d’arracher un électron à celle-ci, qui est ainsi transformée en radical-cation plus ou moins stable. Ce dernier peut rester intact ou bien subir des fragmentations donnant naissance à de nouveaux cations ou groupements neutres de masses plus faibles.Le spectromètre de masse est conçu pour séparer ces fragments suivant leur masse, et pour déterminer celle-ci [cf. SPECTROMÉTRIE DE MASSE].La spectrométrie de masse est une méthode de choix pour déterminer la séquence de peptides de taille faible ou moyenne (au maximum 15 acides aminés): elle est rapide, en grande partie automatique et ne nécessite que de petites quantités de produits (quelques picomoles). Les peptides plus grands et les protéines doivent au préalable être séquencés en fragments plus petits. Afin d’augmenter la volatilité des peptides, les groupements 漣 NH 漣 peptidiques peuvent être transformés en groupements 漣 N 漣 acyl ou 漣 N 漣 CH3 par des méthodes chimiques classiques.La figure 4 représente le spectre de masse d’un octapeptide N-méthylé C6H5 漣CH2 漣 O 漣 C(= O) 漣 Ala 漣 Val 漣 Gly 漣 Leu 漣 Ala 漣 Val 漣 Gly 漣 Leu 漣 OCH3. Le pic à 953 est le pic moléculaire du produit; les pics de masse plus faible correspondent pour la plupart à la perte successive de résidus d’aminoacides à partir de l’extrémité C 漣 terminale: perte de Leu 漣 OCH3, Gly, etc. La détermination de la masse de ces différents fragments permet directement celle de la séquence du peptide.Les techniques récentes de spectrométrie de masse ne nécessitent plus de modifier chimiquement les peptides: il n’est plus indispensable qu’ils soient volatils. L’échantillon est placé dans une matrice de glycérol ou adsorbé sur un support solide. Il est ensuite ionisé par un faisceau à haute énergie d’atomes ou d’ions, comme dans les méthodes du fast atom bombardment (FAB-MS) ou celle de l’émission ionique secondaire (SI-MS). Dans la technique dite par electrospray , une solution de protéine dans un solvant volatil est pulvérisée dans le spectromètre de masse. Sous vide, le solvant des gouttes s’évapore rapidement, laissant les molécules de protéines en suspension. Ces techniques permettent notamment de déterminer le poids moléculaire de protéines jusqu’à 100 000 daltons (environ 1 000 aminoacides) avec une précision de 0,01 p. 100.La spectrométrie de masse est de plus en plus souvent utilisée couplée en série à une première méthode d’analyse, comme dans le cas de la chromatographie en phase gazeuse (GC-MS), de la chromatographie liquide haute pression (HPLC-MS) ou encore de la spectrométrie de masse elle-même (MS-MS). Les constituants d’un mélange complexe peuvent être ainsi directement identifiés.2. Structure des composés minérauxDiffraction des rayons XLa structure cristalline permet non seulement une étude du type de liaison, mais aussi une approche de la formule effective des composés minéraux. Par exemple, le composé Cu3(ZrF7)2, 16 H2O est en réalité formé d’assemblages de trois ions. Le zirconium n’y est pas heptacoordiné comme le ferait penser la formule globale, mais octacoordiné avec un polyèdre de coordination formé d’un antiprisme d’Archimède (fig. 5); le cuivre est hexacoordiné, mais l’un des ions est mononucléaire tandis que l’autre est dinucléaire. La formule s’écrira donc:
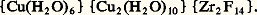 Spectroscopie ultraviolette et visibleLes spectres électroniques des composés des éléments de transition possèdent des caractéristiques liées à l’entourage de l’élément. Soit le cobalt (II). Celui-ci possède sept électrons d (d 7). Un diagramme simplifié des niveaux d’énergie est représenté sur la figure 6. À gauche, on a les états d’énergie de l’ion libre Co2+; à droite, on observe la transformation de ces états sous l’action d’un champ tétraédrique (groupe de symétrie Td en notation due à Schönflies) des coordinats qui l’entourent. Seuls les états les plus importants sont représentés. Trois transitions sont attendues: 42 良 4A2 ( 益1 région 3 000-5 000 cm size=1漣1), 41(F) 良 4A2 ( 益2 région 7 000 cm size=1漣1), 41(P) 良 4A2 ( 益3 bande structurée vers 15 000-20 000 cm size=1漣1). Les spectres d’un groupe de composés pseudo-tétraédriques CoX2(HMTA)2 sont donnés sur la figure 7. On y observe diverses positions des bandes d’absorption suivant la nature de X (X = Cl, Br, I, NCS, NCSe); ces différences sont liées à la «série spectrochimique». Pour une symétrie donnée des coordinats, les états d’un ion libre sont perturbés; les spectres permettront d’en déduire la stéréochimie d’une molécule ou d’un ion. C’est ainsi que les bandes du spectre du sulfate de cobalt hydratéCo(H2O)6S4, H2O ont des positions et des intensités différentes, car le champ des molécules d’eau est octaédrique.Spectroscopie d’absorption atomiqueUn faisceau de lumière monochromatique traverse un nuage d’atomes neutres obtenus à partir d’un échantillon. Ces atomes passent d’un niveau d’énergie à un niveau plus élevé. La différence d’énergie entre les deux niveaux correspond à une radiation 益0 située entre 200 et 900 nm pour un élément métallique. La spectrophotométrie d’absorption permet ainsi de déterminer le nombre d’atomes neutres présents dans un échantillon. Cette méthode est spécifique du métal analysé, ce qui nécessite une source par métal à doser pour éviter toute interférence. Pour obtenir ces atomes neutres, on utilise deux modes d’atomisation: l’atomisation par flammes et l’atomisation thermique sans flamme. Les sensibilités peuvent être de 3 猪g . l size=1漣1 dans le cas du magnésium pour le premier mode cité avec des résultats reproductibles. Elle est de 0,1 猪g . l size=1漣1 pour le même élément dans le second mode. Cette grande sensibilité va cependant de pair avec une moins grande reproductibilité.Cette méthode spécifique et sensible est notamment utilisée dans l’étude des minéraux à l’état de trace dans les eaux.Dispersion rotatoire optiqueL’étude de la dispersion rotatoire optique est de plus en plus utilisée pour de nombreux composés optiquement actifs du cobalt (III), de l’iridium (III), du rhodium (III) de formuleM(AA)3n + (avec AA coordinat bidenté – cf. CHÉLATION).Spectroscopie infrarouge et RamanEn chimie minérale, les vibrations de valence et de déformation sont spécifiques du site occupé par un atome ou un groupe d’atomes non seulement dans les molécules, mais aussi dans les composés qui forment un réseau rigide comme les spinelles et les perowskites. Dans ce dernier cas, la présence du cristal est un inconvénient, car les bandes observées sont larges. Par contre, avec des composés moléculaires, il existera des bandes bien définies, leur emplacement et leur nombre seront liés très nettement au nombre de coordination et à la stéréochimie (tabl. 2). L’étude Raman complémentaire à la spectrophotométrie infrarouge sera efficace si la molécule possède un centre de symétrie. Une vibration active en spectroscopie Raman sera inactive en infrarouge et inversement (cf. effet RAMAN).Résonance magnétique nucléaireLa représentation des signaux observés en R.M.N. de noyaux (fluor, phosphore, vanadium, mercure, etc.) permet de déduire la stéréochimie de composés minéraux. Le nombre de pics dans le spectre d’un atome A est donné par la formule n A = 2 SB + 1. Un atome A subit en effet le couplage d’un atome non équivalent B. La somme des spins des noyaux B est donnée par SB = n BIB, où IB représente le spin nucléaire. On obtient par exemple des molécules pseudo-octaédriques par réaction du tétrafluorure de titane avec des oxydes de pyridine substituée L. Le déplacement chimique lu sur les spectres réalisés à partir de solutions dans l’acétonitrile comme solvant, à 漣 40 0C, permet de savoir si les deux coordinats L fixés au titane (fig. 8) sont placés en cis ou en trans (cf. STÉRÉOCHIMIE). Pour une configuration a , tous les fluors sont identiques et ne sont pas couplés entre eux; on observera un simple pic: c’est le cas avec le coordinat L égal à 2,6 -(CH3)2C5H3NO. Pour une configuration b , il existe deux sortes de fluor qui sont couplées; on observera ainsi deux triplets (L = C5H5NO). Lorsque les deux configurations sont en équilibre, comme avec L représenté par CH3C5H4NO, les deux triplets et le singulet coexisteront.Une autre technique dite de l’Indor (Internuclear Double Resonance ) permet la détermination sans équivoque de la structure dans des composés hétéronucléaires.Résonance paramagnétique électroniqueUne bonne idée des liaisons dans les produits minéraux a été acquise grâce aux études de résonance paramagnétique électronique (R.P.E.) car beaucoup d’ions métalliques sont des radicaux, c’est-à-dire possèdent des électrons célibataires. Les principes de base de cette spectroscopie sont proches de ceux de la spectroscopie R.M.N.; cependant, mis à part quelques cas, les critères d’observation des spectres R.M.N. et R.P.E. sont incompatibles. En R.P.E. il faut que le temps de relaxation électron-spin soit long. Une fréquence micro-onde est absorbée par des ions, des molécules ou des atomes possédant des électrons avec des spins non appariés. La sensibilité des spectromètres est très importante car on peut détecter un oxyde manganeux dans 1 000 oxydes de magnésium. La fréquence de résonance qui se situe dans la gamme des ondes radar est obtenue par un champ magnétique H0 tel que h 益 = g 廓H0. Suivant la fréquence type utilisée, on aura des appareils en bande X (9,5 GHz = 9,5 憐 109Hz) ou en bande Q (35 GHz), c’est-à-dire des longueurs d’onde de l’ordre du centimètre. Des informations sur la nature de l’ion métallique, sur le degré d’oxydation, sur la liaison métal-coordinat, sur la symétrie, la distribution électronique et aussi l’ordre des niveaux d’énergie et sur la force du champ des coordinats dans les composés paramagnétiques peuvent être tirées du spectre. Pour certains noyaux, tels le vanadium ou le niobium, des renseignements sur la structure électronique sont obtenus grâce à l’interaction entre les moments magnétiques des électrons et ceux du noyau. On a alors un couplage hyperfin. Chaque niveau électronique peut être alors décomposé en 2I + 1 niveaux nucléaires pour un spin nucléaire I (fig. 9). Quelquefois les couplages peuvent avoir lieu avec les noyaux des coordinats (1H, 13C, 19F, 31P...), c’est le couplage superhyperfin (fig. 9). Le facteur de décomposition spectrale g dévie par rapport à celui du spin libre (g 0 = 2,0023). Avec quelques éléments de transition, ce facteur g est égal à g 0 漣 n, où est la séparation énergétique entre l’état fondamental et l’état excité, n un nombre entier et le couplage spin-orbite.Une des techniques de double résonance (Indor) augmente la sensibilité et la résolution de cette méthode. Son développement apporte des renseignements sur des couplages à longue distance et la structure spatiale d’une molécule paramagnétique.Résonance quadrupolaire nucléaireLa spectroscopie de résonance quadrupolaire nucléaire (R.Q.N.) a été entrevue lorsque ont été observés des couplages quadrupolaires nucléaires. La grande facilité de détection des fréquences R.Q.N. des halogènes (sauf pour le fluor 19F pour lequel cet effet n’existe pas) a été mise à profit. Par exemple, on peut déduire que le chlorure d’or (III) possède deux sortes d’atomes de chlore. La structure plane de Au2Cl6 contient en effet quatre atomes de chlore en position terminale et deux en position pontée entre deux atomes d’or. L’un des inconvénients est la quantité importante nécessaire aux mesures par rapport aux autres méthodes.Spectroscopie électroniqueLa spectroscopie photoélectronique, ou E.S.C.A. (Electron Spectroscopy for Chemical Analysis ), consiste à étudier la distribution énergétique d’électrons émis par une irradiation de rayons X. Tous les électrons peuvent être extraits. Des photons de grande énergie pénètrent à l’intérieur de la structure électronique. On différencie ainsi un atome, mais aussi un même élément lié dans un composé, car l’énergie cinétique que l’électron possédera à la sortie, pour une même couche quantique, n’est pas la même suivant la liaison de cet atome avec les voisins. On met ainsi en évidence deux sortes d’azote dans un composé tel que(C2H5)4N2Co(NCS)4. L’énergie de liaison de l’électron 1s est différente de celle de l’azote lui-même (fig. 10); elle est plus faible pour l’azote du groupement isothiocyanate et plus forte pour l’azote du cation tétraéthylammonium. On constate aussi qu’il y a plus d’azote dans l’anion que dans le cation. La méthode est si sensible qu’elle permet de différencier le cobalt parmi les 180 autres éléments contenus dans la vitamine B12.La spectroscopie électronique Auger est une méthode directe d’analyse chimique des surfaces. Elle fait intervenir l’ionisation de la couche électronique par des rayons d’énergie de l’ordre de 3 keV et permet par exemple d’identifier du carbone et du soufre. On peut noter que le phénomène Auger a lieu plus particulièrement pour des atomes légers (fig. 11) alors que la fluorescence a lieu pour des atomes plus lourds.Spectroscopie MössbauerEn disposant des atomes radioactifs dans une matrice à l’état solide, on peut obtenir des rayons 塚 sans recul du noyau (cf. effet MÖSSBAUER). Les rayons 塚, d’énergie très élevée, issus d’un noyau excité peuvent rencontrer un noyau identique dans son état fondamental; il est alors réabsorbé. Si la source se déplace par rapport à l’absorbant, on obtient une résonance si l’environnement du noyau n’est pas le même que celui de la source (effet Doppler). Parmi les renseignements obtenus, signalons les effets de covalence, le caractère 神, la valence de l’élément et sa stéréochimie. Beaucoup d’éléments sont concernés; les plus importants sont le fer, l’étain, l’iode et l’europium. La figure 12 représente un effet quadrupolaire résultant de la transition entre un état de spin nucléaire 1/2 et un état supérieur excité de spin 3/2. Ce dernier état présente en effet une distribution de charge non sphérique. Il en résulte un moment quadrupolaire donné par l’écart entre les deux maximums. La figure 13 montre l’existence de deux écarts quadrupolaires 1 et 2 pour des échantillons de formiate ferreux hydraté. On en déduit qu’il existe à l’intérieur du cristal deux sortes de sites, le plus grand écart étant lié au site octaédrique déformé par quatre molécules d’eau.On peut observer aussi des modifications électroniques par variation de la température (fig. 14).
Spectroscopie ultraviolette et visibleLes spectres électroniques des composés des éléments de transition possèdent des caractéristiques liées à l’entourage de l’élément. Soit le cobalt (II). Celui-ci possède sept électrons d (d 7). Un diagramme simplifié des niveaux d’énergie est représenté sur la figure 6. À gauche, on a les états d’énergie de l’ion libre Co2+; à droite, on observe la transformation de ces états sous l’action d’un champ tétraédrique (groupe de symétrie Td en notation due à Schönflies) des coordinats qui l’entourent. Seuls les états les plus importants sont représentés. Trois transitions sont attendues: 42 良 4A2 ( 益1 région 3 000-5 000 cm size=1漣1), 41(F) 良 4A2 ( 益2 région 7 000 cm size=1漣1), 41(P) 良 4A2 ( 益3 bande structurée vers 15 000-20 000 cm size=1漣1). Les spectres d’un groupe de composés pseudo-tétraédriques CoX2(HMTA)2 sont donnés sur la figure 7. On y observe diverses positions des bandes d’absorption suivant la nature de X (X = Cl, Br, I, NCS, NCSe); ces différences sont liées à la «série spectrochimique». Pour une symétrie donnée des coordinats, les états d’un ion libre sont perturbés; les spectres permettront d’en déduire la stéréochimie d’une molécule ou d’un ion. C’est ainsi que les bandes du spectre du sulfate de cobalt hydratéCo(H2O)6S4, H2O ont des positions et des intensités différentes, car le champ des molécules d’eau est octaédrique.Spectroscopie d’absorption atomiqueUn faisceau de lumière monochromatique traverse un nuage d’atomes neutres obtenus à partir d’un échantillon. Ces atomes passent d’un niveau d’énergie à un niveau plus élevé. La différence d’énergie entre les deux niveaux correspond à une radiation 益0 située entre 200 et 900 nm pour un élément métallique. La spectrophotométrie d’absorption permet ainsi de déterminer le nombre d’atomes neutres présents dans un échantillon. Cette méthode est spécifique du métal analysé, ce qui nécessite une source par métal à doser pour éviter toute interférence. Pour obtenir ces atomes neutres, on utilise deux modes d’atomisation: l’atomisation par flammes et l’atomisation thermique sans flamme. Les sensibilités peuvent être de 3 猪g . l size=1漣1 dans le cas du magnésium pour le premier mode cité avec des résultats reproductibles. Elle est de 0,1 猪g . l size=1漣1 pour le même élément dans le second mode. Cette grande sensibilité va cependant de pair avec une moins grande reproductibilité.Cette méthode spécifique et sensible est notamment utilisée dans l’étude des minéraux à l’état de trace dans les eaux.Dispersion rotatoire optiqueL’étude de la dispersion rotatoire optique est de plus en plus utilisée pour de nombreux composés optiquement actifs du cobalt (III), de l’iridium (III), du rhodium (III) de formuleM(AA)3n + (avec AA coordinat bidenté – cf. CHÉLATION).Spectroscopie infrarouge et RamanEn chimie minérale, les vibrations de valence et de déformation sont spécifiques du site occupé par un atome ou un groupe d’atomes non seulement dans les molécules, mais aussi dans les composés qui forment un réseau rigide comme les spinelles et les perowskites. Dans ce dernier cas, la présence du cristal est un inconvénient, car les bandes observées sont larges. Par contre, avec des composés moléculaires, il existera des bandes bien définies, leur emplacement et leur nombre seront liés très nettement au nombre de coordination et à la stéréochimie (tabl. 2). L’étude Raman complémentaire à la spectrophotométrie infrarouge sera efficace si la molécule possède un centre de symétrie. Une vibration active en spectroscopie Raman sera inactive en infrarouge et inversement (cf. effet RAMAN).Résonance magnétique nucléaireLa représentation des signaux observés en R.M.N. de noyaux (fluor, phosphore, vanadium, mercure, etc.) permet de déduire la stéréochimie de composés minéraux. Le nombre de pics dans le spectre d’un atome A est donné par la formule n A = 2 SB + 1. Un atome A subit en effet le couplage d’un atome non équivalent B. La somme des spins des noyaux B est donnée par SB = n BIB, où IB représente le spin nucléaire. On obtient par exemple des molécules pseudo-octaédriques par réaction du tétrafluorure de titane avec des oxydes de pyridine substituée L. Le déplacement chimique lu sur les spectres réalisés à partir de solutions dans l’acétonitrile comme solvant, à 漣 40 0C, permet de savoir si les deux coordinats L fixés au titane (fig. 8) sont placés en cis ou en trans (cf. STÉRÉOCHIMIE). Pour une configuration a , tous les fluors sont identiques et ne sont pas couplés entre eux; on observera un simple pic: c’est le cas avec le coordinat L égal à 2,6 -(CH3)2C5H3NO. Pour une configuration b , il existe deux sortes de fluor qui sont couplées; on observera ainsi deux triplets (L = C5H5NO). Lorsque les deux configurations sont en équilibre, comme avec L représenté par CH3C5H4NO, les deux triplets et le singulet coexisteront.Une autre technique dite de l’Indor (Internuclear Double Resonance ) permet la détermination sans équivoque de la structure dans des composés hétéronucléaires.Résonance paramagnétique électroniqueUne bonne idée des liaisons dans les produits minéraux a été acquise grâce aux études de résonance paramagnétique électronique (R.P.E.) car beaucoup d’ions métalliques sont des radicaux, c’est-à-dire possèdent des électrons célibataires. Les principes de base de cette spectroscopie sont proches de ceux de la spectroscopie R.M.N.; cependant, mis à part quelques cas, les critères d’observation des spectres R.M.N. et R.P.E. sont incompatibles. En R.P.E. il faut que le temps de relaxation électron-spin soit long. Une fréquence micro-onde est absorbée par des ions, des molécules ou des atomes possédant des électrons avec des spins non appariés. La sensibilité des spectromètres est très importante car on peut détecter un oxyde manganeux dans 1 000 oxydes de magnésium. La fréquence de résonance qui se situe dans la gamme des ondes radar est obtenue par un champ magnétique H0 tel que h 益 = g 廓H0. Suivant la fréquence type utilisée, on aura des appareils en bande X (9,5 GHz = 9,5 憐 109Hz) ou en bande Q (35 GHz), c’est-à-dire des longueurs d’onde de l’ordre du centimètre. Des informations sur la nature de l’ion métallique, sur le degré d’oxydation, sur la liaison métal-coordinat, sur la symétrie, la distribution électronique et aussi l’ordre des niveaux d’énergie et sur la force du champ des coordinats dans les composés paramagnétiques peuvent être tirées du spectre. Pour certains noyaux, tels le vanadium ou le niobium, des renseignements sur la structure électronique sont obtenus grâce à l’interaction entre les moments magnétiques des électrons et ceux du noyau. On a alors un couplage hyperfin. Chaque niveau électronique peut être alors décomposé en 2I + 1 niveaux nucléaires pour un spin nucléaire I (fig. 9). Quelquefois les couplages peuvent avoir lieu avec les noyaux des coordinats (1H, 13C, 19F, 31P...), c’est le couplage superhyperfin (fig. 9). Le facteur de décomposition spectrale g dévie par rapport à celui du spin libre (g 0 = 2,0023). Avec quelques éléments de transition, ce facteur g est égal à g 0 漣 n, où est la séparation énergétique entre l’état fondamental et l’état excité, n un nombre entier et le couplage spin-orbite.Une des techniques de double résonance (Indor) augmente la sensibilité et la résolution de cette méthode. Son développement apporte des renseignements sur des couplages à longue distance et la structure spatiale d’une molécule paramagnétique.Résonance quadrupolaire nucléaireLa spectroscopie de résonance quadrupolaire nucléaire (R.Q.N.) a été entrevue lorsque ont été observés des couplages quadrupolaires nucléaires. La grande facilité de détection des fréquences R.Q.N. des halogènes (sauf pour le fluor 19F pour lequel cet effet n’existe pas) a été mise à profit. Par exemple, on peut déduire que le chlorure d’or (III) possède deux sortes d’atomes de chlore. La structure plane de Au2Cl6 contient en effet quatre atomes de chlore en position terminale et deux en position pontée entre deux atomes d’or. L’un des inconvénients est la quantité importante nécessaire aux mesures par rapport aux autres méthodes.Spectroscopie électroniqueLa spectroscopie photoélectronique, ou E.S.C.A. (Electron Spectroscopy for Chemical Analysis ), consiste à étudier la distribution énergétique d’électrons émis par une irradiation de rayons X. Tous les électrons peuvent être extraits. Des photons de grande énergie pénètrent à l’intérieur de la structure électronique. On différencie ainsi un atome, mais aussi un même élément lié dans un composé, car l’énergie cinétique que l’électron possédera à la sortie, pour une même couche quantique, n’est pas la même suivant la liaison de cet atome avec les voisins. On met ainsi en évidence deux sortes d’azote dans un composé tel que(C2H5)4N2Co(NCS)4. L’énergie de liaison de l’électron 1s est différente de celle de l’azote lui-même (fig. 10); elle est plus faible pour l’azote du groupement isothiocyanate et plus forte pour l’azote du cation tétraéthylammonium. On constate aussi qu’il y a plus d’azote dans l’anion que dans le cation. La méthode est si sensible qu’elle permet de différencier le cobalt parmi les 180 autres éléments contenus dans la vitamine B12.La spectroscopie électronique Auger est une méthode directe d’analyse chimique des surfaces. Elle fait intervenir l’ionisation de la couche électronique par des rayons d’énergie de l’ordre de 3 keV et permet par exemple d’identifier du carbone et du soufre. On peut noter que le phénomène Auger a lieu plus particulièrement pour des atomes légers (fig. 11) alors que la fluorescence a lieu pour des atomes plus lourds.Spectroscopie MössbauerEn disposant des atomes radioactifs dans une matrice à l’état solide, on peut obtenir des rayons 塚 sans recul du noyau (cf. effet MÖSSBAUER). Les rayons 塚, d’énergie très élevée, issus d’un noyau excité peuvent rencontrer un noyau identique dans son état fondamental; il est alors réabsorbé. Si la source se déplace par rapport à l’absorbant, on obtient une résonance si l’environnement du noyau n’est pas le même que celui de la source (effet Doppler). Parmi les renseignements obtenus, signalons les effets de covalence, le caractère 神, la valence de l’élément et sa stéréochimie. Beaucoup d’éléments sont concernés; les plus importants sont le fer, l’étain, l’iode et l’europium. La figure 12 représente un effet quadrupolaire résultant de la transition entre un état de spin nucléaire 1/2 et un état supérieur excité de spin 3/2. Ce dernier état présente en effet une distribution de charge non sphérique. Il en résulte un moment quadrupolaire donné par l’écart entre les deux maximums. La figure 13 montre l’existence de deux écarts quadrupolaires 1 et 2 pour des échantillons de formiate ferreux hydraté. On en déduit qu’il existe à l’intérieur du cristal deux sortes de sites, le plus grand écart étant lié au site octaédrique déformé par quatre molécules d’eau.On peut observer aussi des modifications électroniques par variation de la température (fig. 14).
Encyclopédie Universelle. 2012.